Importing external mutations¶
For instructions on installation and basic exploration steps, see Getting started.
It is possible to post-process lists of mutations with isomut2py that were otherwise generated with an external variant caller tool. Here we demonstrate how external VCF files can be loaded and further analysed.
As isomut2py expects pandas DataFrames as inputs to handle mutation lists, we will be using the pandas package for the loading of VCF files.
[2]:
import isomut2py as im2
import pandas as pd
%matplotlib inline
Compiling C scripts...
Done.
Importing VCF files using pyvcf¶
VCF files can be easily processed in python with the pyvcf package. This can be installed with:
pip install pyvcf
[7]:
import vcf
To convert VCF files to pandas DataFrames with columns that can be parsed by isomut2py, we first need to make sure to find the values of sample_name, chr, pos, type, score, ref, mut, cov, mut_freq, cleanliness and ploidy. In order to perform downstream analysis of mutations, fields cov, mut_freq and cleanliness can be left empty, but nonetheless have to be defined in the dataframe.
Here we are importing VCF files generated by the tool MuTect2 (of GATK). If another tool was used for variant calling, make sure to modify parser function below. The example VCF files are located at [exampleDataDir]/isomut2py_example_dataset/ExternalMutations/mutect2. (For instructions on how to download the example datafiles, see Getting started.)
[91]:
def parse_VCF_record(record):
if record.is_deletion:
muttype = 'DEL'
elif record.is_indel:
muttype = 'INS'
elif record.is_snp:
muttype = 'SNV'
return (record.CHROM, record.POS, muttype,
record.INFO['TLOD'][0],
record.REF, record.ALT[0],
int(record.samples[0].data.DP),
int(record.samples[0].data.AD[1])/int(record.samples[0].data.DP),
int(record.samples[1].data.AD[1])/int(record.samples[1].data.DP))
[108]:
sample_dataframes = []
for i in range(1,7):
vcf_reader = vcf.Reader(filename = exampleDataDir + 'isomut2py_example_dataset/ExternalMutations/mutect2/'+str(i)+'_somatic_m.vcf.gz')
d = []
for record in vcf_reader:
d.append(parse_VCF_record(record))
df = pd.DataFrame(d, columns=['chr', 'pos', 'type', 'score', 'ref', 'mut', 'cov', 'mut_freq', 'cleanliness'])
df['sample_name'] = 'sample_'+str(i)
df['ploidy'] = 2
sample_dataframes.append(df[['sample_name','chr', 'pos', 'type', 'score', 'ref',
'mut', 'cov', 'mut_freq', 'cleanliness', 'ploidy']])
mutations_dataframe = pd.concat(sample_dataframes)
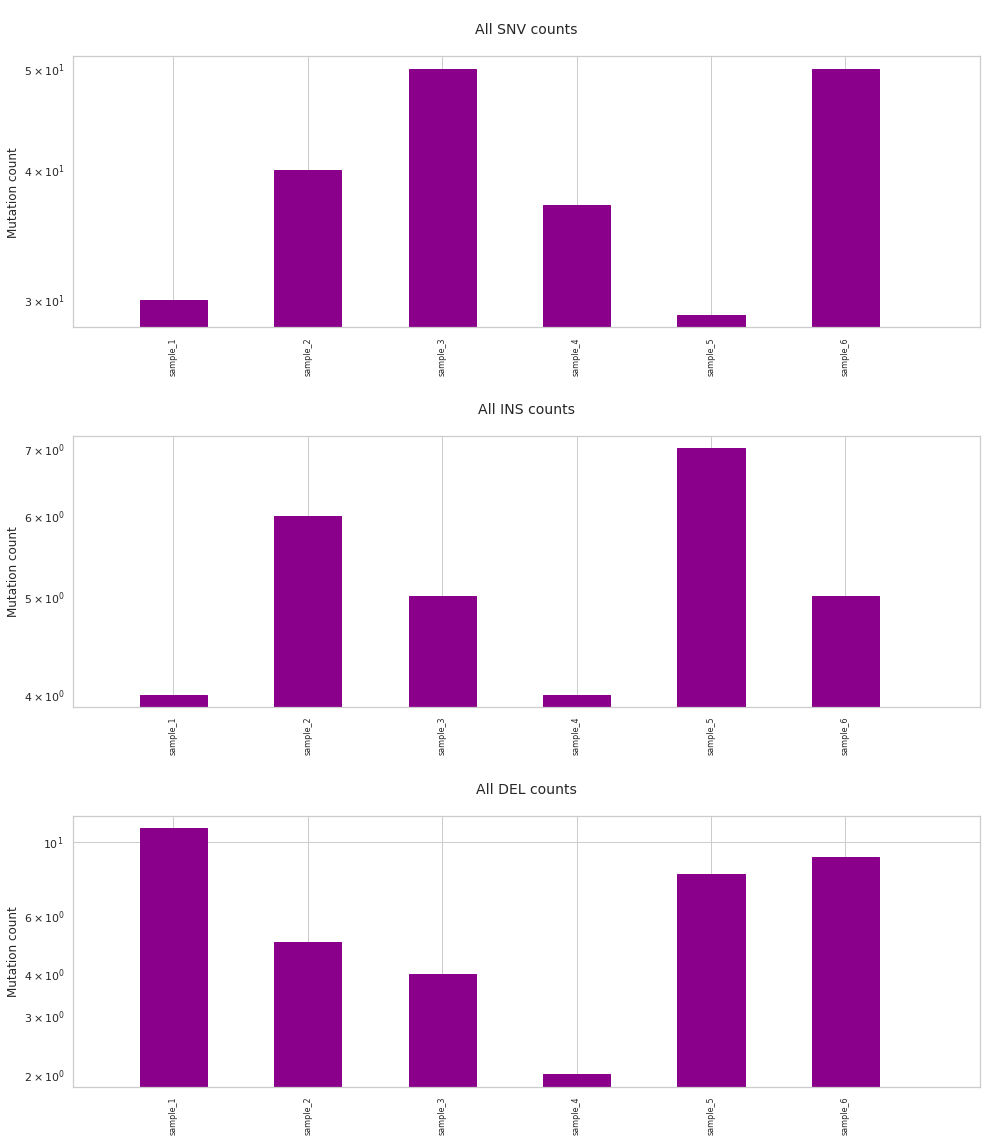
Now we have all the mutations found in 6 samples in a single dataframe. This can be further analysed with the functions described in Further analysis, visualization. For example the number of mutations found in each sample can be plotted with:
[112]:
f = im2.plot.plot_mutation_counts(mutations_dataframe=mutations_dataframe)
Warning: list of control samples not defined.
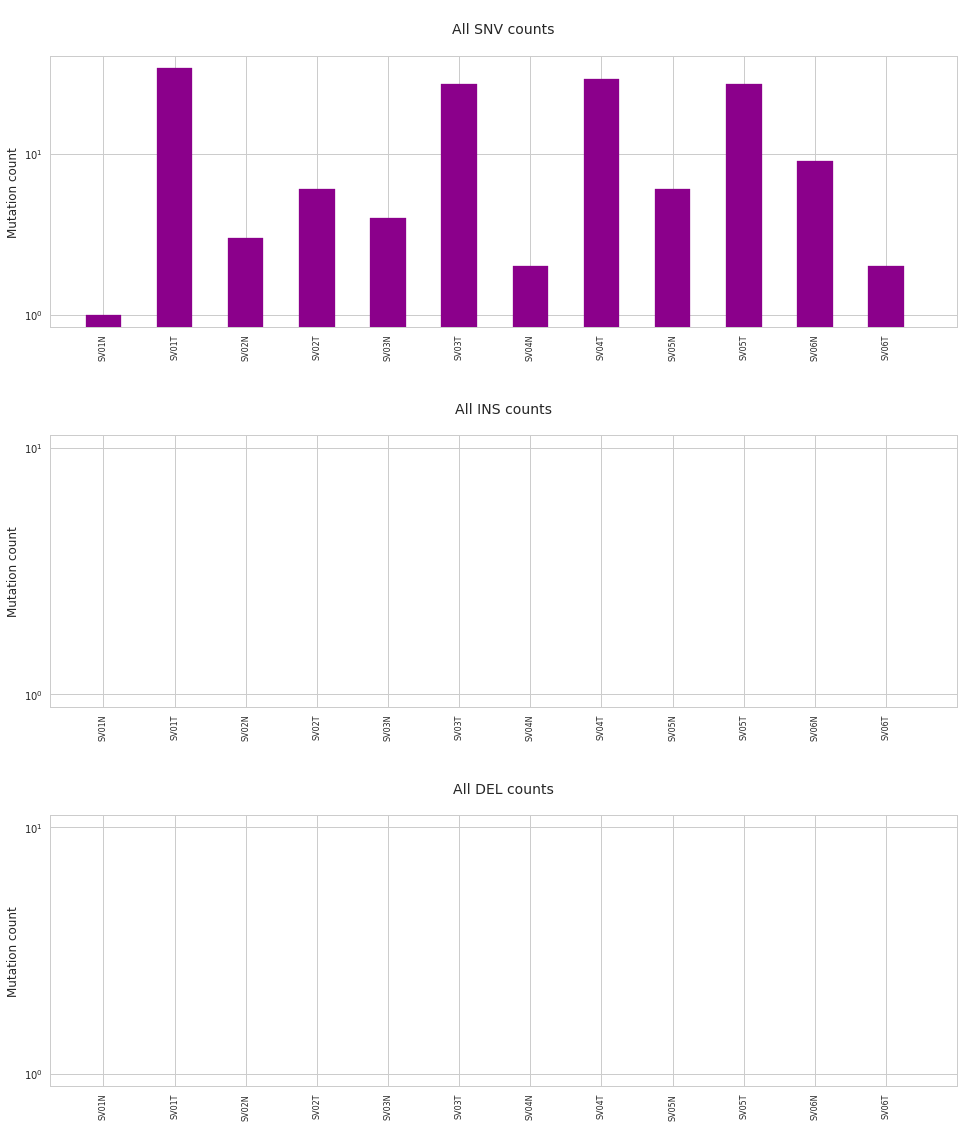
Importing lists of mutations in arbitrary files¶
If your lists of mutations are stored in some kind of text files as tables, the easiest way to import them is to use the pandas python package. (As isomut2py heavily relies on pandas, it should already be installed on your computer.)
Here we merely include an example for such an import, make sure to modify the code and customize it to your table. (This data was generated with MuTect, that only detects SNPs, thus we don’t expect to see any indels.)
[4]:
exampleDataDir = '/nagyvinyok/adat83/sotejedlik/orsi/'
[27]:
sample_dataframes = []
for i in range(1,7):
for sample_type in ['N', 'T']:
df_sample = pd.read_csv(exampleDataDir + 'isomut2py_example_dataset/ExternalMutations/mutect/SV0'+
str(i)+sample_type+'_chr1.vcf', sep='\t', comment='#')
df_sample = df_sample[df_sample['judgement'] != 'REJECT']
df = pd.DataFrame()
df['chr'] = df_sample['contig']
df['pos'] = df_sample['position']
df['type'] = 'SNV'
df['score'] = df_sample['t_lod_fstar']
df['ref'] = df_sample['ref_allele']
df['mut'] = df_sample['alt_allele']
df['cov'] = df_sample['t_ref_count'] + df_sample['t_alt_count']
df['mut_freq'] = df_sample['t_alt_count']/df['cov']
df['cleanliness'] = df_sample['n_ref_count']/(df_sample['n_ref_count'] + df_sample['n_alt_count'])
df['ploidy'] = 2
df['sample_name'] = 'SV0' + str(i) + sample_type
df = df[['sample_name',
'chr',
'pos',
'type',
'score',
'ref',
'mut',
'cov',
'mut_freq',
'cleanliness',
'ploidy']]
sample_dataframes.append(df)
mutations_dataframe = pd.concat(sample_dataframes)
[30]:
import warnings
warnings.filterwarnings("ignore")
[31]:
f = im2.plot.plot_mutation_counts(mutations_dataframe=mutations_dataframe)
Warning: list of control samples not defined.